
Pharmakokinetik beschreibt, was der Körper mit einem Wirkstoff macht. Sie analysiert, wie schnell und in welchem Ausmaß ein Medikament aufgenommen, verteilt, verstoffwechselt und ausgeschieden wird. Gemeinsam bestimmen diese Prozesse, wie stark und wie lange ein Arzneimittel wirkt. Die Pharmakokinetik bildet damit eine zentrale Grundlage für sichere Dosierungen, klinische Studien und die individualisierte Therapie.
Was bedeutet Pharmakokinetik
Pharmakokinetik umfasst die zeitlichen Veränderungen der Wirkstoffkonzentration im Körper. Sie untersucht die vier zentralen Prozesse Absorption, Distribution, Metabolismus und Exkretion. Zusammen erklären sie, wie viel eines Wirkstoffs am Wirkort ankommt, wie lange er dort wirkt und wann der Körper ihn wieder entfernt.
Ein Beispiel verdeutlicht dieses Konzept. Eine Patientin nimmt eine Tablette Ibuprofen ein. Zunächst löst sich die Tablette im Magen auf und der Wirkstoff gelangt in den Dünndarm. Dort beginnt die Aufnahme in den Blutkreislauf. Sobald Ibuprofen im Blut zirkuliert, verteilt es sich im Körper und erreicht die Bereiche, in denen es seine schmerzlindernde Wirkung entfaltet. Parallel beginnt der Körper mit dem Abbau in der Leber und anschließend mit der Ausscheidung über die Nieren.
Dieser Ablauf zeigt, dass nicht allein die Dosis entscheidet, sondern die gesamte pharmakokinetische Reise.
Pharmakokinetik einfach erklärt
Die vier Prozesse lassen sich unter dem Kürzel ADME zusammenfassen. Sie beschreiben den Weg eines Wirkstoffs vom Einnahmezeitpunkt bis zur Ausscheidung. Individuelle Eigenschaften wie Alter, genetische Faktoren, Körpergewicht oder Organfunktion beeinflussen jeden dieser Schritte und damit auch die optimale Dosis.
Die vier zentralen Prozesse der Pharmakokinetik (ADME)
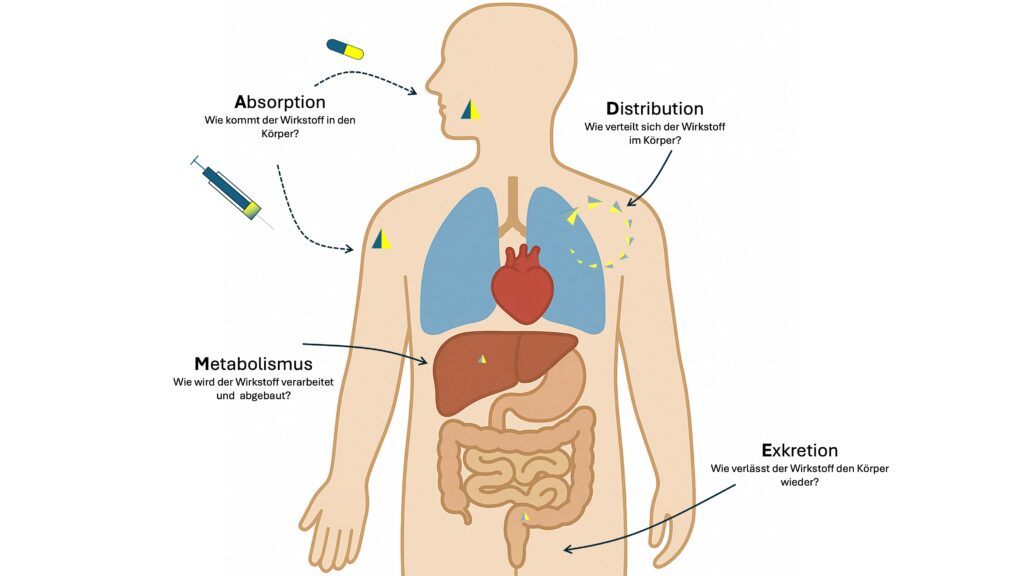
Absorption
Absorption beschreibt die Aufnahme eines Wirkstoffs in den Blutkreislauf. Je nach Darreichungsform verläuft dieser Prozess unterschiedlich. Orale Medikamente passieren den Magen und gelangen in den Dünndarm, wo der größte Teil der Aufnahme erfolgt. Injektionen umgehen diesen Weg und gelangen unmittelbar oder nahezu unmittelbar in den systemischen Kreislauf.
Die klinische Relevanz zeigt sich am Beispiel eines oralen Antibiotikums. Bei starkem Durchfall kann die Darmpassage beschleunigt sein, was die Absorption verringert. Die Folge können unzureichende Wirkstoffspiegel und eine verminderte Wirksamkeit sein.
Distribution
Die Distribution beschreibt die Verteilung des Wirkstoffs im Körper. Entscheidende Faktoren sind Blutvolumen, Gewebeperfusion und Proteinbindung. Nur der freie, nicht an Plasmaproteine gebundene Anteil ist pharmakologisch aktiv.
Ein klassisches Beispiel ist Warfarin. Bei Patienten mit niedriger Albuminkonzentration steigt der freie Wirkstoffanteil im Plasma. Dies erhöht das Risiko für Blutungen und erfordert eine engmaschige Überwachung.
Metabolismus
Der Metabolismus umfasst die Umwandlung eines Wirkstoffs in aktive oder inaktive Metabolite. Die Leber spielt dabei eine zentrale Rolle, wobei Enzyme wie CYP3A4, CYP2D6 oder CYP2C9 entscheidend sind.
Codein dient als typisches Beispiel. Es ist ein Prodrug und wird erst durch CYP2D6 in Morphin umgewandelt. Genetische Varianten führen dazu, dass manche Menschen Codein sehr schnell oder sehr langsam metabolisieren. Die Folge können entweder starke Wirkungen oder nahezu fehlende Effekte sein. Die genetische Variabilität zeigt, wie stark individuelle Faktoren die Pharmakokinetik beeinflussen.
Exkretion
Die Exkretion beschreibt die Entfernung des Wirkstoffs oder seiner Metabolite aus dem Körper. Die Nieren sind dafür meist das wichtigste Organ. Einige Substanzen verlassen den Körper aber auch über die Galle, die Atemluft oder den Schweiß.
Bei eingeschränkter Nierenfunktion können Wirkstoffe wie Aminoglykoside im Körper akkumulieren. Dadurch steigt das Risiko für Nebenwirkungen, weshalb die Medizin die Dosis anhand der glomerulären Filtrationsrate anpasst.
Hinweis: Der Prozess der Liberation
In manchen Darstellungen ergänzt man ADME zu L-ADME. Liberation beschreibt die Freisetzung eines Wirkstoffs aus der Darreichungsform, zum Beispiel aus Tabletten, Kapseln oder Retardpräparaten. Dieser Schritt beeinflusst Geschwindigkeit und Ausmaß der Absorption. Bei intravenösen Injektionen entfällt der Prozess. In der klinischen Praxis konzentriert man sich daher häufig auf die vier Kernprozesse Absorption, Distribution, Metabolismus und Exkretion.
Wichtige pharmakokinetische Parameter
Pharmakokinetische Parameter beschreiben, wie sich Wirkstoffkonzentrationen über die Zeit verändern. Dazu nutzt man meist Blutproben, da sich aus der Konzentrations-Zeit-Kurve wesentliche Parameter wie Cmax, AUC oder die Halbwertszeit ableiten lassen. Je nach Fragestellung können jedoch auch Urin, Stuhl, Atemluft oder Speichel ergänzende Informationen liefern, etwa über renale Eliminierung oder enterohepatische Kreisläufe.
Wie sich diese Konzentrationsverläufe in eine pharmakologische Wirkung übersetzen, ist Gegenstand der PK/PD-Modellierung, bei der pharmakokinetische Parameter mit pharmakodynamischen Effekten verknüpft werden.
Standardparameter im Überblick
- Cmax und Tmax: Cmax beschreibt die maximale Plasmakonzentration. Tmax gibt an, wann diese erreicht wird. Beide Werte zeigen, wie schnell ein Arzneimittel wirkt und helfen dabei, das Risiko für Nebenwirkungen einzuschätzen.
- AUC: Die Fläche unter der Konzentrationskurve zeigt, wie viel eines Wirkstoffs über die Zeit im Körper verfügbar ist. Die AUC ist besonders wichtig, wenn man die Bioäquivalenz von Medikamenten beurteilt.
- Clearance: Clearance beschreibt, wie viel Blut pro Zeiteinheit von einem Wirkstoff befreit wird. Eine niedrige Clearance kann zu einer Anreicherung führen und erfordert häufig eine Dosisanpassung, insbesondere bei eingeschränkter Leber- oder Nierenfunktion.
- Halbwertszeit: Die Halbwertszeit gibt an, wie lange der Körper benötigt, um die Konzentration eines Wirkstoffs zu halbieren. Sie bestimmt maßgeblich das Dosierungsintervall. Substanzen mit langer Halbwertszeit, wie Amiodaron, verbleiben lange im Körper und akkumulieren entsprechend.
- Verteilungsvolumen: Das Verteilungsvolumen beschreibt, wie weit sich ein Wirkstoff im Körper ausbreitet. Lipophile Stoffe weisen oft hohe Verteilungsvolumina auf und verteilen sich tief in das Gewebe.
Erweiterte pharmakokinetische Parameter
| Parameter | Bedeutung | Klinische Relevanz anhand eines Beispiels |
|---|---|---|
| Bioverfügbarkeit (F) | Anteil des Wirkstoffs, der systemisch verfügbar wird | Orales Morphin besitzt deutlich geringere Bioverfügbarkeit als die intravenöse Form, was eine höhere orale Dosis nötig macht |
| Absorptionsratekonstante (ka) | Geschwindigkeit der Aufnahme | Schnell wirksame Analgetika haben ein hohes ka, wodurch die Wirkung rasch einsetzt |
| Eliminationsratekonstante (ke) | Geschwindigkeit, mit der der Körper einen Stoff entfernt | Enzyminduktion erhöht ke und führt zu schnellerer Eliminierung, was höhere Dosen erforderlich machen kann |
| Proteinbindung | Anteil des gebundenen Wirkstoffs im Plasma | Sinkt das Albumin, steigt der freie Anteil von Warfarin und damit das Risiko für Blutungen |
| Steady State (Css) | Gleichgewicht zwischen Aufnahme und Eliminierung | Dauertherapien erreichen nach etwa vier bis fünf Halbwertszeiten den Steady State, zum Beispiel bei Antidepressiva |
| Akkumulationsfaktor (R) | Maß für Anreicherung bei wiederholter Gabe | Amiodaron akkumuliert stark aufgrund seiner sehr langen Halbwertszeit |
| First-Pass-Effekt | Metabolisierung vor Eintritt in den systemischen Kreislauf | Propranolol wird stark präsystemisch metabolisiert, daher sind orale Dosen wesentlich höher als i.v. |
| Extraktionsverhältnis (E) | Anteil des Wirkstoffs, der bei Passage eines Organs eliminiert wird | Lidocain besitzt ein hohes hepatisches Extraktionsverhältnis und ist dadurch flusslimitiert |
| Intrinsische Clearance | Metabolisierungsfähigkeit eines Organs unabhängig vom Blutfluss | Bei schwerer Leberinsuffizienz sinkt die intrinsische Clearance deutlich |
| Peak-to-Trough-Ratio | Verhältnis zwischen Spitzen- und Talspiegel | Vancomycin erfordert ein enges Monitoring, um hohe Peaks und zu niedrige Troughs zu vermeiden |
| Fluktuationsindex | Schwankungen zwischen zwei Gaben | Retardpräparate zeigen geringere Schwankungen und werden oft besser vertragen |
| Absorptionslagzeit (tlag) | Verzögerung bis zum Beginn der Absorption | Retardtabletten zeigen häufig eine tlag, weshalb der Wirkungseintritt verzögert ist |
Pharmakokinetische Modellierung und Anwendung
Pharmakokinetische Modelle helfen dabei, Konzentrationsverläufe zu verstehen und Dosierungen gezielt zu steuern. Viele Parameter lassen sich nicht direkt messen, sondern basieren auf modellgestützten Schätzungen.
Mathematische Modelle
Bei der Auswertung pharmakokinetischer Daten verwendet man häufig Ein- oder Mehrkompartimentmodelle. Sie beschreiben, wie sich ein Wirkstoff zunächst im Blut verteilt und anschließend in verschiedene Gewebe übergeht. Damit diese Modelle realistische Konzentrationsverläufe abbilden, nutzen sie nicht nur die gemessenen Blutspiegel, sondern auch physiologische Informationen aus Vorstudien. Dazu gehören Organvolumina, Blutflussraten, Proteinbindungsdaten, die Lipophilie des Wirkstoffs sowie Erkenntnisse aus präklinischen Verteilungsstudien. Aus der Kombination dieser Daten schätzen die Modelle Parameter wie Eliminationsratekonstanten, Verteilungsvolumina oder Absorptionsgeschwindigkeiten, die sich aus Konzentrationsmessungen allein nicht ableiten lassen. Auf diese Weise entsteht ein mathematisches Abbild des Stoffwechsels, das individuelle Unterschiede sichtbar macht und die Grundlage moderner Dosierungsstrategien bildet.
Klinische Anwendung
Pharmakokinetische Modelle unterstützen vielfältige Entscheidungen im therapeutischen Alltag und in klinischen Studien. Sie helfen nicht nur dabei, die geeignete Dosishöhe festzulegen, sondern auch das optimale Dosierungsintervall zu bestimmen. Dadurch lässt sich planen, wie häufig ein Medikament verabreicht werden muss, um wirksame und zugleich sichere Konzentrationen zu erreichen. Gleichzeitig liefern die Modelle Hinweise darauf, zu welchem Zeitpunkt eine Gabe besonders sinnvoll ist, etwa ob ein Arzneimittel morgens, abends oder in Bezug auf Mahlzeiten eingenommen werden sollte. Auf diese Weise ermöglichen pharmakokinetische Informationen eine individuelle Anpassung verschiedener Dosierungsschemata, unabhängig davon, ob es um orale Gaben, Bolusinjektionen, Infusionen oder retardierte Präparate geht.
Darüber hinaus ist die Pharmakokinetik in allen Phasen der Arzneimittelentwicklung relevant. In frühen Studien bewertet sie die Wirkstoffexposition und unterstützt die Dosisfindung. In späteren Phasen zeigt sie, ob therapeutisch wirksame Konzentrationen erreicht werden und wie individuelle Unterschiede die Therapie beeinflussen. Das Therapeutische Drug Monitoring nutzt pharmakokinetische Modelle zudem, um Dosierungen präzise an einzelne Patienten anzupassen. Ein klinisches Beispiel ist Vancomycin. Zu niedrige Spiegel vermindern die Wirksamkeit, während zu hohe Spiegel das Risiko für Nierenschäden erhöhen. Moderne Leitlinien berücksichtigen dafür zunehmend AUC-basierte Zielbereiche.
Fazit
Pharmakokinetik beschreibt den Weg eines Wirkstoffs durch den Körper und erklärt, wie Absorption, Distribution, Metabolismus und Exkretion die Wirkung eines Arzneimittels bestimmen. Ein fundiertes Verständnis dieser Prozesse ist entscheidend für die Entwicklung neuer Medikamente, die Planung klinischer Studien und die sichere Anwendung im medizinischen Alltag.
Weiterführende Literatur:
Ernstmeyer, K., & Christman, E. (Hrsg.). (2023). Pharmacokinetics. In Nursing Pharmacology (2. Aufl., S. 21–48). https://www.ncbi.nlm.nih.gov/books/NBK595006/